- Jan 4, 2025
Basics of the Common Technical Document (CTD)
- Caroline Ritchie
- 0 comments
What is the Common Technical Document (CTD)?
The CTD is a standardized format used to organize and submit information to regulatory authorities for drug approval and lifecycle management. It was developed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) to streamline and harmonize the drug submission process across regions, primarily the United States, Europe, and Japan.
See the ICH website for more information.
How is the CTD Organized?
The CTD is divided into 5 modules, each with specific information:
-
Module 1: Administrative and Product Information
Specific to the region where the submission is being made.
Includes forms, application cover letters, labeling, and regional-specific requirements.
Note: Module 1 is often NOT considered part of the CTD because it is not “common” across regions, but it is still an important component of regulatory submissions.
-
Module 2: Summaries
High-level summaries of the scientific information in Modules 3, 4, and 5.
Includes a Quality Overall Summary, Nonclinical Overview, Clinical Overview, and associated summaries.
-
Module 3: Quality
Detailed information about the drug's chemistry, manufacturing, and controls (CMC).
Covers drug substance and drug product development, manufacturing processes, specifications, and stability data.
-
Module 4: Nonclinical Study Reports
Data from preclinical studies, such as pharmacology, toxicology, and ADME (absorption, distribution, metabolism, excretion) studies.
-
Module 5: Clinical Study Reports
Data from clinical trials, including efficacy and safety data, statistical analyses, and clinical trial summaries.
The CTD is often represented as a triangle, with Modules 3, 4, and 5 forming the base.
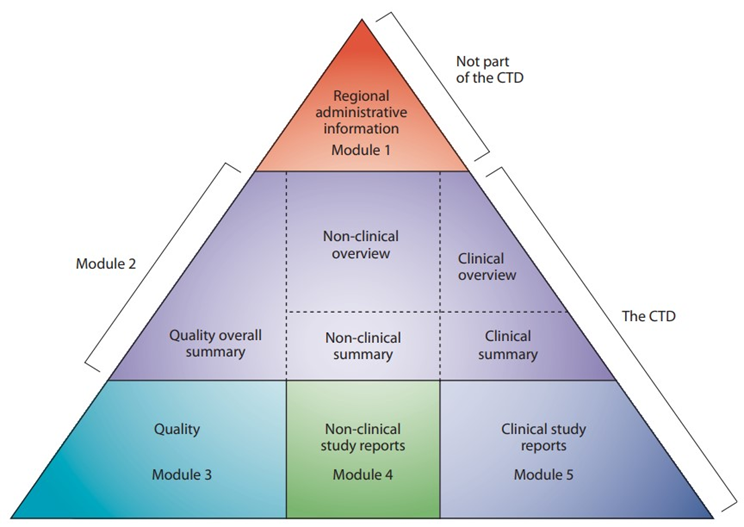
Source: https://www.ich.org/page/ctd
The CTD serves as the format for submitting a marketing authorization application (MAA) for new drugs or biologics to health authorities including:
The US Food and Drug Administration (FDA)
The European Medicines Agency (EMA)
Japan’s Pharmaceuticals and Medical Devices Agency (PMDA)
Note that while the FDA, EMA, and PMDA were the founding members of the ICH, many other health authorities have adopted ICH standards and accept submissions using the CTD framework.
The detailed granular structure of the CTD is described in ICH M4: Organization of the Common Technical Document for the Registration of Pharmaceuticals for Human Use. There are individual ICH guidance documents for certain parts of the CTD. For example, M4E describes the clinical components of the CTD, while M4S describes the nonclinical components of the CTD. All of these guidance documents, along with questions and answers documents, can be found here.
Having a single format for an MAA across multiple regions ensures a consistent structure across regions, making it easier for companies to prepare submissions for multiple markets with minimal redundancy and for reviewers to navigate data. This saves companies time and money and speeds up the submission and approval process across multiple regions.
When is the CTD Submitted?
The CTD is typically submitted during:
New Drug Application (NDA) or Biologics License Application (BLA) in the US
Marketing Authorization Application (MAA) in Europe
It may also be used for supplemental applications, variations, and periodic updates
Note that an Investigational New Drug (IND) Application in the US can serve as the foundation of the CTD, as some content required for an IND will also be used or updated for an NDA/BLA. Therefore, content submitted for an IND typically follows the CTD structure.
Real-World Representation of the CTD
I always think of the CTD as a large filing cabinet with a ton of folders. When an entity with therapeutic potential (a small molecule or biologic) is identified, a pharmaceutical company starts populating some of the empty folders in one of these filing cabinets. As more information is gleaned from the development process, more of the folders are filled with information. Once sufficient information is obtained (and folders are filled) to file an IND, the company may decide to do so. Once the IND is cleared, clinical studies will progress and, eventually, more folders will be filled. Eventually, sufficient information is obtained to file an NDA/BLA (or MAA in other regions). Each of these “filings” or “submissions” is sent to health authorities in the CTD format – meaning that the file cabinet is sent to a health authority. The file cabinet of every entity with therapeutic potential from every company is organized in the same way but not every folder will be filled consistently (for example, some subfolders may not be required for some entities and some entities will have more clinical trials than others). For submission in different ICH regions, the Module 1 folders would simply be swapped out for each region.
How is the CTD Submitted?
An entire CTD for an MAA could be well over 100,000 pages. In decades past, companies would literally send moving trucks filled with multiple paper copies of their MAA documentation to the FDA or another health authority.
Most regulators now require submissions in electronic format (ie, the electronic CTD or eCTD). This allows efficient review, tracking, and lifecycle management of applications. Fancy software programs allow for building entire submissions using the CTD framework, allow documents to talk to each other (eg, hyperlinks can be added across documents, not only within documents), and allow submission through each health authority’s electronic gateway, allowing secure transmission and validation of submissions.
With the eCTD, the entire submission can be transmitted with the click of a button. Not only is this convenient for the companies who prepared the MAA, but it has changed how documents within a submission are prepared. Documents within the CTD can now cross-reference other documents within the CTD, minimizing redundancy and saving reviewers a lot of time by allowing access to pertinent information with the click of their mouse.
Summary
Anyone supporting a regulatory submission must understand the structure and purpose of the CTD. It is the mechanism by which data and information are submitted to health authorities. It is the bridge between the drug development process and regulatory approval.
In summary, the CTD format simplifies the regulatory submission process by:
Facilitating global harmonization and quicker access to multiple markets.
Enhancing the efficiency of regulatory review.
Reducing duplication of efforts for multinational submissions.